Unknown
0 views18289 WordsCopy TextShare
Unknown
Video Transcript:
so just going to make a few introductory points here I wanted to mention that we are here in large part because the doctor to be well and his lab we are going to be talking about things related to our knee seek but in the context of those things that were pointing out by him and his collaborators that are important right now the presenters are going to be dr. Adam Eric golonka he's the chief scientist at the tower bioinformatics research center and he's going to be presenting together with Julia panov she's a PhD student and both
of them will share both biological interpretation or biological projects and some technical things those work is supported by the Taliban from Attucks Research Center which is that University of Haifa in Israel and also Pine biotech when we are going to be talking about some data examples based on data these are published datasets we provide here links to those published data sets you're welcome to read the publication's and also download data it's available and also I want to point out that some of these projects are prepared as educational project project and are available on our platform
our platform is the first of all the website that you're on right now edu dot t-- - bio info and you will find there are also some courses on basics of data analysis similar to what you're going to be seeing here today so just an overview of what we will cover today or hope to cover first of all it's going to be about next-generation sequencing data pre-processing including trimming technical sequences and PCR duplicates and the second point about processing of next-generation sequencing is going to be around quantification of expression levels conventional pipelines and also identification
eisah forms we will touch on the challenge of gene set enrichment analysis so annotation of genes but also gene set enrichment and finally we will practice practice and utilize some machine learning methods including unsupervised analysis of expression data and supervisor now so now I'm going to hand over the presentation to Julia and Julia will share with you her presentation about biological examples ok should be able to share your screen now ok hi can you see my screen can you see my screen yes okay great so hi everyone Azealia mentioned my name is Julia and I
used the platform in my research for a neurodevelopmental disorders and I also collaborate and assist other groups in using the platform in their research in areas such as marine biology evolutionary biology and cancer studies but I'm actually on the wrong slide right yeah you want me to show your slacks okay Julia I have your slides here if you want to I'm trying to get this working I'm sorry for some hopefully sorry so okay so my goal today before we begin the hands-on part of the workshop is to give you a taste of what we can
do with the top robotics platform and also with bioinformatics in general so I will focus on one project that we have been working on and I will briefly tell you about the kind of data and the analysis we used to generate the results that I will present afterwards every step of the analysis that you will be hearing about was done on the platform so let me begin yeah I'm on the wrong slide I'm not in the present mode wait oh yeah yes sorry about this but my sharing screen is not working for me could you
do it for me and share my and share the presentation for me and I will just speak okay can you hear or can you see my screen yeah okay great so and so maybe you can show the next slide so I will focus on one project about cell lines and I will tell you about the kind of data and the analysis that we use to generate the results that I present afterwards and so let me begin the project we will be looking at is based on the analysis by the gray group and I hope you
had a chance to to look at the publication but if you didn't it's okay we will be I will go as well so the 70 cell lines of breast cancer and we used only 49 of these cell lines that had all the molecular profiles for them and as you can see on the slide we had gene expression data and RNA editing data so the RNA seek data is actually produced by sequencing the RNA molecules in the sample so from this RNA sequencing we can get two kinds of data we can get the gene expression data
that we will go into more detail and you will have a chance yourself to practice how to do it exactly and also we can do RNA editing and we could generate the RNA editing table from those data as well so what it what edit those are might've forgotten or don't know is a post-transcriptional modification of primary RNA molecules as you can see on the slide sometimes RNA molecules are by a special enzyme and there are different types of editing there is a substitution of cytosine to uracil that in the sequencing data we see as a
timing and another type of modification could be a substitution of adenine to by go online so we studied these two modifications together with gene expression and RNA editing events can have very big effects on afterwards untranslated protein and the protein can be unfunctional or it can lose the biting sites for micro rna's that it had in wild-type state and the exact mechanism and the function of editing events are not completely understood but it has been shown that RNA editing plays significant role in different types this is about the kind of data we were looking at
gene expression and RNA editing and question is why why would someone might be interested in data driven analysis that actually has no planning beforehand we were not looking at specific genes or perturb rated States we were just looking at the data overall so we were interested actually in trying to identifies subtypes of breast cancer as you know breast cancer is a heterogeneous disease and our understanding of its complexity is still evolving so on this slide you can see kind of our history of what it was how people have with time have been trying to identify
the subtypes of cancer breast disease and recently people are trying to identify subtypes of cancer based on what we call multi-omics analysis by different layers RNA data DNA data data all together one analysis to find specific unique characteristic Euler characteristics of subtypes of cancer and as you probably know one of the methods or model cancer our cell lines and these cell lines are subtypes of cancer or or types of cancer and they are well established cell lines originally taken from patients that underwent biopsies and they are very useful models for studying the cancer as you
can see on the next slide we can have profiles of different types of cell lines with genomic profile and as well as pharmacological profile and based on these profiles we can better find treatments for patients that have similar genomic profile so of course cell lines are not are not the best models of cancer they lack some of the important things that cancer and patients have and that is hydrogen media inside the tumor cells and also the stromal component and however despite the limitations cell lines continue to be very important research models and preclinical trial tools
in cancer biology so understanding the characteristics of different cell lines can help us better understand both the subtypes of cancer and also sometimes understand that some cancer line is not a good model for the forecast for the type of cancer that it was used for as a motto so in our on the next slide you will see that our our project was based on 49 such cell lines we had gene expression table and you can see on the slide the gene expression table is actually a collection of all genes in stem we have different samples
with different profiles for each gene so expression and on the bottom table RNA editing table you see chromosome regions of the genome that have RNA editing events with high probability and what we're trying to do on the next slide you will see that to take these two tables and find groups of genes that are and groups of RNA editing that together have the similar sample profiles so you can see on the slide that for example we could find a group of genes in yellow and a group of RNA editing events also in yellow and they
would give us clustering of samples that are very similar to each other you can see the purple arrows pointing to the clustering of the samples and and those genes and RNA editing events would be somehow linked or another case could be that the genes in green and RNA editing events in green that could also give us a separation of samples a different separate example or for example an outlier sample that is somehow unique in its molecular characteristics and so the goal of our analysis in this in in this part in this project was exactly that
to find these somehow different samples that are different in molecular characteristics from all others and so this is actually what we were able to do was to identify one unique cell line and that was very different from all others on the next slide you will see matrixes of distances between samples so this is those are distances between samples and you can see in blue there are lines in blue on both tables one table is distances of samples based on the gene expression table and on the other table take on the other side is the matrix
of distances between samples based on the RNA editing events and you can see that both of these tables actually separate only one cell line HCC 202 you can see that separation in blue the distances between HCC 202 and all other cell cell lines are very high and they are painted in blue for visual representation so another way of showing this is going to be on the next slide is to look at the pca over the actual if you took took now these genes and you would look at the pca based on the expression level of
these genes you would see that these genes that we chose are only present in this sample and also with RNA editing events they are also only present in this signal so without actually designing our experiments so that we could target specific genes we were still able to choose a group of genes and a group of RNA editing events that clearly separates this sample from all others so let's look a little bit at kind of genes these are on one side we found olfactory receptors and micro rna's those were the expression levels and on the other
side we found rabjeet a basis so can we from knowing these genes can we now say anything about this cell line how is it different from all others well we could have some hypothesis and afterwards we could maybe generate more experiments or take more data to validate our hypothesis one of the hypotheses we were thinking of is that micro RNAs are known to bind to genes and to prevent the expression of these genes micro RNAs are also known to bind to a RAB gtp aces which are very important transport molecules and when Rob gypped a
gtpases are edited sometimes these binding sites are missing and no more no longer micro RNAs can be buying it can bind to this to these genes because of that we would have a very high expression of tranport not like us trap databases and it's known in the cancer studies that all factory receptors can cause malignancy in cancer or aggressiveness in cancer so wrap gtp aces by trafficking the olfactory receptors to nearby cells can actually make the cancer cell line or the cancer tumor much more aggressive so this would be some kind of hypothesis that could
be validated more with more targeted with more targeted experiments later on so this was a brief glimpse at what can be done with data driven by informatics approach and we hope that now you have a little bit of a more understanding of why this approach can be helpful in biological studies and life scientists life sciences and now I hope you will enjoy the work the hands-on workshop so thank you very much for your attention okay Julia and now I'm going to switch it over to Vladimir as it was said before glad in there is the
chief scientist at the table bioinformatics research center he as working development of the platform and extending it for both research and education [Music] a microphone isn't a zoo okay okay so hope that the screen is shared correctly if there are any problem please let me know and before we start please let us check that all of you have an access to your accounts at the tab whereby in the platform and that you have all the files in the archive prepared for this workshop so please let us know if anyone does not have this files as
we'll use them later on in our practice so in any case we begin with some theory and then we'll proceed to practical analysis hopefully the theory will be relatively short and it will be related to the basis of RNA sequencing pipeline I mean library preparation that we need the step that we need to understand in order to understand what are the main steps of the analysis as you know perfectly well everything starts with the genome which part codes proteins so genes code for proteins but proteins are not generated just directly from genome there's an intermediate
step genes give rise to transcripts to my to messenger rna's and these messenger RNAs are then translated to proteins and all the cell's for the fixed organized for example all the cell's for one human well generally if we avoid thinking about cancer cells for a while generally have the same genes but these genes have different activity different levels of activity different expression levels in different cells in different tissues so some genes may be present in the genome but give rise to no messenger RNA is no transcripts and hence no proteins some can give rise to
an essential amount of transcripts and essential thus essential amount of proteins and roughly speaking sequencing sequencing process can be described as follows we have all these transcripts these transcripts are randomly divided into small pieces and this pieces are read by sequencing machine so read letter by letter nucleotide by nucleotide the problem is that we do not know the origin for all these pieces who get after this reading but anyway we know the sequence for human genome we know the nucleotide sequences for all the genes so we can map our shout segments on transcripts or better
to say probably better to say on genes and if there are no reads mapped on a gene for example gene bet shean be we see that this gene has zero or at least low expression level more reads more short segments we have the higher the expression level is but if we go deeper in the details we'll see that this process has several intermediate steps these steps might be slightly different between technologies but generally these steps are the following first all the transcripts are shattered into sub pieces so we have shattering and in fact we have
one copy of each segment more copies of the transcript of a transcript we have more pieces related to this gene we're served in our sample of the chattering at the next step adapters are negated to these small pieces in fact these prayer reads in fact this Legation is successful not for all small pieces but at least these adapters are gated to a sufficient part of this short reads them PCR amplification is performed that is multiple copies of each read with legate adapters is generated for example if there are six round of PCR amplification well if
there are two rounds of PCR amplification we get to multiple by two four copies for each piece if we have six rounds of PCR amplification we get to power 6 so 64 copies more rounds we have we get more copies for each segment there is a slight problem related to the fact that this higher efficiency is different for different segments so for some pieces we have more copies for some pieces we have lower numbers of copies so we're speaking about average numbers of copies web gain at this step so please pay attention to the fact
that indeed there are multiple copies of each segment after this step but sequencing takes not all the pieces but takes only a relatively small fraction of these pieces and reads these small fraction if the number of copies is relatively low it's the case when the initial number the initial amount of RNA is sufficient and the quality of library preparation is also sufficient then generally will read only one copy for each short segment number of duplicates so number of reads originated from the same small piece of initial transcript will be well above well below 10% if
we start with lower amount of RNA if so we need to perform more steps more cycles of PCR amplification we get more copies for each segment and the number of these VCR duplicates increases more over the absence of uniformity and the number of copies starts to play the role but after that we can continue to our previous discussion we simply map all these reads on initial genes and estimated an expression level just counting reads mapped on each gene so this logic this logic explains us this logic explain us the major steps of the analysis we
may start with pre-processing so if there are additional segments negated additional sequences negated to our initial segments adapters this adapters may be removed prior to the analysis additionally there's a standard effect of low quality amps for reads they also can be trimmed to increase mapping quality then PCR duplicates so identical reads that are potentially the corpus of the same initial segment can be removed from the analysis this steps increase the quality of the resultant mapping so we map reads on known genes unknown transcripts but at this stage there are different strategy different strategies we may
focus just on estimation on expression level of known genes in this case we map all this reads to the set of non genes known transcripts known isoforms we can also want to try to identify novel transcripts novel isoforms of known genes or completely novel transcripts it's a more rare type of the analysis but it's also not technically hard and definitely there's a combined strategy after the reads are mapped we can proceed to the quantification of expression level still just in order to understand how it can be performed technically without going deep into the details of
mapping algorithms of the estimation algorithms let us get started with the use of the terrible Barre info platform let us prepare the pipeline that performs all these jobs we've discussed we won't run this pipeline as the size of sequencing data is generally relatively large so processing time is also relatively large it may take hours or even several days if we have many samples so we'll just prepare these pipeline in order to do it we proceed to the platform please let us know if there are any problem with it and we go to the analysis related
to RNA sequencing data so here it is so yes there's introduction so what we can choose here what are the options today who are dealing with human genome so we select the set of genes of the genome as you see so we select Homo sapiens there are two types of reads single end and pair and for example for our today discussion we have some parent data and we need to okay I think I see it from here great thank you so let's just try okay and the size of the size of sequencing data is large
so uploading of new files can also be slow we use just links to the files that are already inside the system to have this short test so plug in here somebody was just asking if I think just to make sure so he'll is everybody on the same page can we move forward or okay if you go back one slide okay currently I'm just change screen so if again we are inside the platform so when we log in we can press on the areas of the analysis link and then we've got sections for analysis of various
types of omics data and for various pre-processing here we go to the analysis of RNA sequencing data so here's the link on the top left-top link so here we're here in order to perform the analysis we to perform standard analysis we use type of sequencing data we have single end sequencing or parent sequencing well currently in many cases its parent PE parent and select genome or the set of genes select transcriptome we're working is so parent and Homo sapiens and then we upload files so we press on upload files and add groups button please let
me know if there are any problem with what's going on and for our case for the case when we use files that are already inside we use URL bulk upload here it is so here we go to the files in the archive you have main files and for example if you want to start with just two samples we use the file links two samples dot spld this is a very small file which contains only links to our data but not the data itself okay so dick leagues if you do not have these files please let
us know and we'll simply fix this problem there was a question so what are the four drop-down options so okay okay so first we can deal with different types of experimental data I mean model organisms organism for each genome and transcriptome is known so and we have everything known for an organism our human sapiens so is the first option data type we're dealing is so typically for RNA sequencing we start with first few files if we begin from the very beginning then the type of reads single and or pair and and if we're dealing with
model organisms we just select genomic and transcriptomic data for related to this organism so organism and rich type so then okay okay then we upload a file from our set okay just to clarify about our set what what we mean is there is a button to download files on the page that you're on so you should have downloaded the zip file and that zip file contains multiple folders one of the folders is called main files web will mainly deal with main files so you download the file you unzip the file inside you see the folder
called main files and then we will be using the files that have an extension dot svl so okay and we select this file hopefully we'll get used to it promptly as we'll create a number of pipeline today of pipelines today the file is links to samples and then we go further with our analysis here we're not going to make differential expression analysis or any complicated analysis currently were focused on just estimation of expression level so after pressing define groups we simply say that we need no groups no additional information for this analysis so cancel no
groups and then we start creating our pipeline so the pipeline should be started so again I select start then a possible option is okay here again so let me just show it once again here we use bulk uploads as we're uploading only links to files but not the files themselves so later on we'll use main uploading button add files but currently were using bulk upload button and upload the file links to samples dot svl so we then press on define groups and say that we have no groups and go on so okay let's take a
look whether they're right and a problem at this stage if not we'll try to go further okay okay so we initialize our pipeline with the start module start and okay then for trimming adapters and low-quality ends of reads we can for example use trim emetic module we press on the module and then confirm by pressing save PCR duplicate removal can be performed where the module PCR clean so default option default technical option is perfectly okay mapping can be performed if we're going to map all our reads on jeans on transcriptome for example with bowtie or
bwh for example with bowtie and expression level quantification can be performed there are some expression table module here we need to choose the units for expression level quantification we'll discuss this units in a minute or two so for example fragments per kilobytes per million map reads it became will also do and that's all that's all so this model will give us quantifications for expression levels here we can just finish our pipeline and model and save give a name to this pipeline for example test pipeline or whatever you like and then in order to run pipeline
we won't do it now as this pipeline takes okay okay so now we won't run this by plan we just looked at how this pipeline can be created so while we're trying to solve our problem let me just repeat all these steps some people are saying that they are still doing making the pipeline so yeah please let me know when everything is okay I'll just repeat all the steps for people for going on just to make visual what we're doing so I just recreate this pipeline I start with the areas of the analysis RNA sequencing
select parent sequencing select genome upload our file links to samples tell that I need no groups for the analysis then start for example dream adapters if I select a model which is not required I selected traumatic but I want to remove it from the pipeline I can press just right mouse button and remove it so removing is performed with the right mouse button but I add traumatic I add PCR cleaning I use mapping this time for example bowtie I after mapping I need to go from reads from mapped reads from read positions to expression level
quantification which is before and by or same expression table and that's all and that is all and pipeline name and in order to start this pipeline I need to press run pipeline but currently I won't do it okay so is everything okay with this simple test we'll okay okay so anyway it's just an example and as you see we can easily include different types of models for each step in our analysis for example we want we can I'm just going on with my order because is not essential things I just gives several comments for people
or already who have already created these pipeline so there are different moods for example adding sequence adding mapping on genome and cufflinks calf merge models will also allow us to add steps related to the identification of neural isoforms so algorithmically it's not so simple but as we have all these modules all these algorithms inside the platform the usage is really simple we add additional modules to the pipeline and wait for the result more modules more execution time but definitely it's not a problem for the majority of experiments when the data is collected for weeks months
and sometimes even for years so additional data to of the analysis is sometimes not essential but from the other hand faster analysis is definitely what who also wants to help blue diamond yeah there are some questions that Sahil wants to ask on behalf of some people [Music] ok so what I understood the question is what do these buttons actually do to explain a little bit more in detail what traumatic and DCR clean you do so that it's clear to everyone it's not just buttons on the screen ok so thank you very much for your question
and let us return to our standard scheme of the analysis of RNA sequencing which just followed our general idea of the experimental of the preparation of experimental data so first in we located the adapters to our short sequences and standard pipeline may may wants to remove these short sequences this is performed by the first model we added traumatic so traumatic loops at the ends of reads tries to identify these standard adapters and has this model also has functionality to try to identify normal adapters which are not in its database forms just trimming so remove these
adapters from the reeds we have sequenced then we discussed these are duplicates we discussed PCR duplicates and so our next step was to remove these PCR duplicates and this is what is done by the is our clean algorithm so it just looks for pairs triples quadruples and so on of identical reads and leaves only one copy so the forms these bizarre duplicate removal the next step is mapping these short reads on genes so for each gene and for each short segment we want to find out whether this read has a good position above these gene
so and after this stage we've got positions on the transcription set for each read several some reads can have now allowed positions and for some reads we may fail to understand to identify its origin several some reads can be mapped on different locations but all this information is obtained by the mapping model for example as we are making on the transcriptomic data bowtie bwa sounds like that and the final step is going from read counts to to go from this mapping to the identification of expression levels to the quantification of expression levels and it's performed
by them and it's performed by the our same expression table modules so definitely algorithms that light behind these titles are not very simple especially if we're speaking about mapping and quantification but the purpose of these models is just as I described so let us please now have a very short pause I suppose for three or four minutes just a short technical break and after this break we'll try to discuss what are the standard units of quantification why we should go from raw read counts to some more complex measures so currently just for a moment just
for several me okay can everyone hear me okay during this break it would be helpful I assumed that everybody had a chance to take a look at the first pipeline that we created let me turn on my video here okay okay so while we're taking this short little break I'm going to turn on the microphone for Co and and please the heel if you want to just make sure that anyone has questions [Music] sorry I could not hear that cou yeah so we you know you have the quality of weeds and I think we can
touch on that subject but in the meantime what I want to do in this short break is just maybe collect all of the questions and so we will be able to answer them one by one so we noted it down the question number one is quality of raw fast Q files before you start the mapping [Music] [Music] [Music] by using this pipeline okay about the eyes it forms just a quick note we will get to that great question I think as Louie has shown with in the beginning we had a method to combine multiple sources
with Muhammad's data identify relationships between he knows I think we will get to that form again another question of minutes artistic apps so the basis of the connections like I have person it means to have these happen that app whether I will be okay so what is that I what is the logic behind selecting specific steps okay so there are different methods that the form of the task generally I hope that we discussed the logic of the preparation of the experimental data and so here here each step of the analysis simply follows the experimental step
just in two or three minutes I'll tell you that in reality several steps can be emitted for example pre-processing can be emitted but generally this pipeline just use back from reads we have out of the sequencing machine to initial data we edit adapters has remove them we potentially edit be shared duplicates had hands we remove them and in fact go back to the initial set of short sequencing sequences this reads originated from original transcripts then we try to map all these short sequences on genes in order to understand which gene which genes are associated with
many segments these genes are the genes with high expression level which genes have no almost no short segments associated with this gene so these genes are genes with low expression so unfortunately if we now go my deeper into the details will fail to complete even a short overview because indeed this app reasons have been studies designed for I think more than at least fifteen years and there are great algorithmic ideas behind this algorithm but currently we're focus just on overview these steps okay questions yes and just maybe you can quickly help address those so the
question number one was how to know quality of raw fast queue files before the mapping question number two was about the logic of different steps of analysis question number three was about making correlations between multiple omics data question number four was about identification of isoforms so I know you probably don't want to address everything at the same time but maybe you can just give an overview of when those questions okay so starting from the beginning we can use we can still use pre-processing steps and look at the output statistics of what what we're doing with
these models provide us a statistics for example with the ratio of P shard of reads identified as PCR duplicates and has the reads which have removed we see the number of reads for which adapters has been have been treated but anyway we can there are ways currently not implemented as modules as we use them really to look just at mean base quality reported by the sequencing machine so fast queue files along with a nucleotide for each position contains an estimate of equality for this position and just look at a number of positions with high quality
and low quality to the number of breeds containing only high quality basis it's possible but another possibility is to get statistics after the mapping for example to get number of reads or percentage of reads successfully mapped on genes to get ratio to get proportion of reads that had a unique location with respect to genes and to make a conclusion about the quality of this fescue file based on these overall statistics so prime primarily number of PCR duplicates that were removed number of reads that were not mapped that were mapped uniquely and that were mapped simultaneously
and on different locations so this is a short well not complete answer to the first question so going further about well for example identification of novel isoforms when we try to identify novel isoforms we are looking at reads mapping them not on transcriptome but on genome or better to say on exotic segments of the genome and these reads can link one exam to another exam so reads can start inside one exam and have there are the end of this read in the Artic it's on so this read just generates a link between these exams and
we have a map of all possible links so of all exon exon links supported by reads having this map we can try to identify genes so combinations of exams corresponding to a fix gene or we can find reads well logically let me avoid explaining here the details well fairly well mapped on a part of a genome that is not currently identified identified as known coding region so it's the logic behind identification of novel isoform so i think these are just short answers to two of the four questions well let me avoid discussing links between multiple
omics types today as it's really along topic so Julia just mentioned that there are some in-house tools we have for these analysis for example the Association there are some publicly available tools for example for multi-omics clustering these tools are also collected certain tools are also collected and the platform so there are multi-omics analysis sections well it's these are just some possibilities some are tommix clustering some associations and a number of additional models but it's just a topic for another discussion unfortunately I'm afraid it will take long even to make an introduction here and there was
the fourth question so could you please remind it okay if there are more questions I can also maybe say that we will have a number of breaks further on and it would be probably helpful if we can move on with the material so that we can do some practical aspects of building the pipelines and understanding their logic and as we get to the next stage we can take some more questions there okay fine so apparently first I want to say that there are at least there are two main types of bizarre duplicates artificial posad replicates
which originate from these step of library preparation PCR amplification but there may also be natural TCR duplicates so while performing these pre-processing step removing of these are duplicates you definitely should keep in mind the details of your experiments primarily well as one of the major parameters the amount of RNA material you started with enhance the number of the share implication cycles you performed and if you're especially if you are dealing potentially with short sequences for example not genes but you have focused on micro rna's then these beasts are the shared applicant removal step should be
used with caution or even completely devoted there are some sources so here you may find links that state that these pre-processing steps mister duplicate removal adapter removal well generally improve the quality of the final expression level quantification this improvement is moderate so pre-processing can be even avoided with getting good results so let us currently try to discuss what can what measures for expression level quantification can be used and the first measure we already discussed is very simple it is number of reads mapped on these gene first we can work with just raw counts the total
number of reads mapped on a gene a slight problem here is that several that some reads in many cases amount of reads are mapped simultaneously on different locations so we may want to split this one count count associated with one read between all the locations to which this read has been mapped the simplest way is to split this one count uniformly but in many cases more complex statistical models can be used and for example these are same module we added to the pipeline uses a relatively complex bias and approach so the idea is that we
can perform this splitting of counts associated with the same read in a more sensible way but in any way if we're dealing with counts either raw or so processed so called expected counts after this splitting what we have we may have two genes and for one gene will have many reads mapped of this gene and for another gene we may have much lower of reads mapped on this gene can we say that well natural expression level of the first gene is for example expression level measured in the number of mRNA molecules associated with a gene
can we say that for for the first gene this expression level is higher as we have higher number of reads unfortunately no and the reason is very simple if read if gene is longer so then shattering this gene or mRNA molecular associated in two parts will simply give us more pieces so longer mRNAs give more pieces in comparison with short messenger rna's but the number of molecules we have is the same so in order to get while meaningful quantification we should keep in mind the length of a gene and we may perform norman with respect
to this length of gene another idea behind scaling behind norman is the following imagine that we have one sample we sequence this sample and with them repeat sequencing just the same sample with adding one additional cycle with adding just depth of sequencing i mean reading generating twice as much reads so generally will have two fold increase in the number of reads mapped to each genes simply as we have more reads into consideration more genes in our collection but it's just the same sample and it's natural to have the same estimations of expression levels that is
why we should also keep in mind sequence in depth for example measured in the number of reads mapped on transcripts and that's why we use another scaling item total number of mapped reads and it's just a very standard unit for expression level quantification fragments per kilo base per million mapped reads so the number of reads is scaled by gene or transcript length and by the total number of reads we have another usual unit for expression level quantification is TPM transcripts per million CPM units right for fixed sample only for fixed sample all right simply proportional
to F pkms but the coefficient of the coefficient see here the coefficient of proportionality is selected in such a way that the sum of all TPS for a given sample is exactly is exactly 1 million so let us try to briefly discuss the logic which stands behind this unit to p.m. as unlike fpkm CPM have a very national biological interpretation so let us consider the following amount number of reads mapped on one gene multipled by the mean read length and divided by gene length so let us try to discuss what is the sense behind these
formula number of reads mapped on one gene multiple by meme read length so what we have in the denominator here so what we have here number of reads multiple by mean read length is in fact the number of sequenced nucleotides associated with this gene so number of nucleotides associated with this gene we have after sequencing after the division after dividing this product by gene length we instead get number of molecules number of messenger RNA is associated with this gene or with this either form that we sequenced if we sum these quantities over all genes we
have in fact we simply get the total number of molecules of messenger RNA of the sequencing and if we take this amount this amount for one gene and then divided by the total amount we have a fraction of messenger rna's of transcripts associated with the gene of interest in the health eternity of sequenced molecules so it's just a fraction multiplication by 1 million gives us scaling so CPM tells us the following in each million of messenger rna's in the sample CPM is the amount of molecules associated with the genes who are looking at so for
example if CPM is 10 then out of each million molecules approximately 10 are molecules associated with these gene and another question related to quantification of expression levels is scaled all these quantities read counts fpkm TPM are the quantities measured in so called linear scale so original scale for the majority for the for many types of the analysis this amounts this linear scale amounts are lot scaled so we take logarithm the reason there are multiple reasons for these transformation for example one of the main reasons is related to the statistics as for the majority of genes
the distribution of expression level is law normal so after taking logarithm we get normal distribution which is very good for the analysis in terms of classical probability theory and mathematical statistics another reason is that linear scale is simply similar for dealing with for biology relative differences in expression level are more meaningful than absolute differences if we say that the difference between expression levels is 100 TPMS it will say almost nothing but if we say that the expression level increased twofold threefold tenfold it gives more information and log scaling simply moves these relative differences to differences
that can be obtained by taking standard arithmetic means and differences so for example in log scale two-fold difference is simply a difference by 1 lakh to unit difference equal to 3 lakh fold units is equivalent to the difference to the 8 fold difference in the initial scale the problem with going to the innate to the logarithm scale very well not essential problem frankly speaking is that logarithm is defined only for numbers greater than 0 and for many genes the expression level is just 0 so we can take logarithm we get minus infinity that's why a
small shift is introduced the alias of shift fry and one of the standard one of the standard values is simply 1 1 of possible values but it's not the only option in any case this shift the differences in this shift effect only genes with low expression level and we should also we should keep in mind that low expressed genes that for low expressed genes quantification just experimentally is an exact so we can't be sure that expression level of low expressed gene is measured is measured really precisely that is why genes with low expression level in
many cases are simply removed from the analysis but log scaling is also a standard step of the analysis so running the pipeline we have just writing the pipeline we discussed simply gives us estimates of expression levels in the units we set for the samples we upload it to the platform in order to see results and in order to compare the results for different types of pre-processing including or not including PCR duplicates removal including or not including trimming adapters you may look now won't do it add the files you have in the subfolder output files there's
the next level subfolder earrin a sequence in comparison with an excel file so for two samples here you have just F pkms linear scale it becames for these two samples just without any processing without with pre-processing that includes only adaptive streaming and low quality and streaming and with the prep recession we discussed so shortly if we compare results for the case is no pre-processing versus no duplicates removal or even for the other case you see that for the majority of genes gene expression estimates are roughly the same so for example for these genes along the
x-axis we have expression level quantification obtained by one type of pre-processing and y-axis gives expression level estimate for the other type of pre-processing so for the majority of genes we get the same or approximately the same results but there are some transcripts for which the difference is really high it seems for example that the point that the dot I'm pointing at is not very far from the line x equals y but the difference is more than one and this difference of more than one is a two-fold difference as we're dealing with log to scale so
mass data experiment gives us the general view but for the individual genes for the individual players we have we need additional verification we need additional validation if we want to deal further with some small subset of genes of isoforms identified at this mass experiment so in any case this analysis simply gives us huge tables so it gives us dozens of thousands of numbers for each sample if we perform mapping on only unknown transcripts we have these numbers only for known ones if we add if we add models for the identification of potential novel transcripts that
definitely need additional what level validation then we have estimates as you hear transcripts marked with X log beginning these numbers additionally for potential novel transcripts but the question is what we can do further with this amount of data these expression level tables are not the end of the story they are just the beginning of the story and so now we'll just start discussing what we can do with these data so let us make a decision do we need a break here or yeah please look for that knowledge now in a bathroom or a brief question
straight okay oh okay okay so currently we'll just try to see what we can do with the table with these tables we'll use real tables for the cell line experimental data Julia briefly described out of these dozens of thousands of genes we removed okay okay so break shut break okay well I just noticed that the microphone actually doesn't work that great when you take it far away is it I know it's fine but I think the farther you go from the computer [Music] okay so let us know when we should start again there was a
request from the audience to see the speaker on video especially when there are questions so we are answering to those requests okay so it's a heel back there okay so don't start again please continue okay okay so currently we'll try to see what we can do with these expression tables and we start with a very simple analysis so we have several about 50 cell lines all cell lions originated from patients with breast cancer and it's probable that all these lines are similar to each other as we're dealing with sea lions associated with just the same
pathology but how can we know if it's true or not it's really impossible to go through all these numbers just by eyes and our visualization stops as an dimension equal to two or three so if we have two attributes we can visualize our data as dots in a plane three attributes can provide us Association of samples with dots in a 3d space but we have thousands of genes thousands tens of thousands of genes and so this visualization is looks impossible however we can try to find an optimum plane that keeps as much data as possible
and still allows us provides us an opportunity of visual inspection of the data so currently I'll try to explain what I mean using a very simple example and then we'll return to the presentation so we have a simple Pam so in fact what you see in your screen what you see in your screen is a 2d projection of a 3d object if we choose a plane of projection in a poor way we have very poor understanding of an object we're dealing with so we have just the same object what we can cannot understand what we're
dealing with in this projection change in the plane changing the projection gives us better and better way of seeing these objects for example in this projection you can really understand what is the edge what is the object okay okay so if there are any problem with video please let us know I think it is working it's working okay great so and a very simple thing can be applied to the data we're dealing with we can try to find optimal plane or optimal 3d space or optimal planes that can this help us in visualization of the
data of the data we have so let us try to test the thing I'm talking about so let us create and run our pipeline and expect inspect the result so we'll be working with unsupervised analysis as in these analysis we know nothing about data underlying our samples we think we start at least with the understanding that all samples are similar they originated from the same pathology so in order to perform this simple kind of the analysis we select unsupervised analysis section upload expression table for the analysis this is the file cell line expression data dot
txt we'll take a look inside this file in a minute or two so we select cell lines expression data file okay so main files in the folder we are working with today and the file cell lines expression data healthy great we select this file cell lines expression data txt and press open button so this file is uploaded to the system unlike the previous analysis here we indeed take the file from your computer's to the system so it may take several seconds please let us know in case of any problem so again we add files file
cell lines expression data so that's it and go on continue continue start then our principle component analysis module is PC draw as we want to have plots as an output PC draw the default options here are okay in order to get additional help regarding these options if you need to if you need explanation you can just point on I zooms and have short comments but here we say that one visualization associated with three first principal components we need no scaling centering and transpose explains which type which information gives the arrows and we wish their columns
samples and genes or issac formers expression so he were then it's just the end of our pipeline and pipeline name see draw for example say the name is the name L then after the name is saved simply run this pipeline by pressing run pipeline button that's it each other pipeline is starting it may take several seconds once the pipeline is started even monitor its progress here are in my pipelines pipelines reports the results and everything for all pipelines you previously had so again pressing on the name brings us to the screen with the pipeline progress
while the pipeline runs let us take a look at the file we uploaded in order to understand what we have inside our system what is this file it is a very simple file in which for each sample each cell line and for each jeans out of well about 7,000 jeans we have expression here expression is measured in log scale log two scales fpkm so we use F became units with log two scaling and so initially each cell line is a point in a space with a number of dimensions close to 7,000 well I reload it's
to see their progress so for me almost completed and in order to understand the simple picture we project all these data we project all this data simply to a 2d plane if you low the page you'll see the progress it won't take too long but it may require several dozens of seconds or a couple of minutes to complete in this platform mode you will also get an email notification after the pipeline is finished okay for me it's done so let us additionally wait some time to reloading updates the progress indicator and after the pipeline is
completed you see 100% done signature in the top right corner so if you have the pipeline completed you may take a look at the output the main output of the pipeline is stored in the download pipeline output files folder so in the left folder you see here download pipeline output files these are the main output files so go in here for example we have a PDF file PC plots so this is the visualization we wanted to have so I download this file and open it each dot is associated with one sample and we started our
discussion from the fact that all cell lines originated from the patients with the same pathology and we wanted to see whether all cell lines are similar however here we see at least two groups of samples we definitely see a clear separation of samples into two groups probably somebody will see additional separation in two groups in the left or in the right parts at the same time we know that there are multiple subtypes of breast cancer and who may hypothesize that this separation is related to the subdivision into subtypes and the authors of this study we
are working with the author of the study that produced the data classified all the samples into four classes into four types of the breast cancer luminal subtype basal subtype cloud in low subtype and normal like subtype so in fact we have information about clinical subtype for each sample and probably it would be interesting to see how subtypes are located into in these plots well we can describe the picture but a better way to look at it is to generate the picture in the same folder in the main file folder we have an additional file expression
data macht it is just the same file but for which each sample is marked by a subgroup is marked by subgroup so let us repeat generating PCA PCA plot but upload this file cell lines expression days marked in order to get different colors for dots associated with different subtypes so the analysis is just the same but the picture will include information about the predefined class so in order to do it we create one more by plying just the same one we used unsupervised analysis in this unsupervised analysis we upload the file but this time we
upload the file expression data marked [Music] we upload the file expression data marked that's it so again the difference is that just here we have predefined classes for samples and we'll see how these classes are located in planes and all the same steps are the following is a simple analysis so we upload the data we upload the data and then just repeat the steps we did continue part piecing you draw for example with different with default parameters and give a name to the pipeline and run it okay maybe this is a good time to see
if everybody is following along and if there's any questions that we have I have this especially the gene names with this or the data needs to be in this particular format of having samples versus did you get that Redeemer Oh could you please repeat the question because the order is a bit worse than I want it to be yes I understand the question is about formatting the input file for the PCA how do you have to organize your transcripts and samples very much perfect question so here we have multiple options we can first for the
PCA for the principle component analysis transport or gene IDs are simply ignored so in this type of the analysis we have features but feature names are not essential then we can arrange samples by columns and features by rows or vice versa and all we need let me show you I'll just show you it on the screen I upload well any file all we need here is to tell system which goes by rows and we what information goes by columns in our case when samples are columns we said transpose equal to yes and in the opposite
case we simply said transpose equal to no and if we have additional information about classes or groups or something like that we simply add a line marked by a keyword group here to here the keyword is groups with a name of a group so again here are two options are possible what goes by rows what goes by columns both options are possible okay are there any other questions so meanwhile I've got a question to you so if we look at the previous PC what here it is on my screen and remember that there are four
subtypes what's your idea how these subtypes would be located two on the left and two on the right or something different so can we make a prognosis looking at this picture - just get feeling the hill can you be the voice of the feeling and so what does the voice of the feeling say so frankly speaking I also have no hypothesis looking at this figure but anyway let's look at the correct answer so hopefully the pipeline is completed for you as well so please let me know if I'm wrong for the next pipeline will simply
have a loop you will simply have a look at new plots we have here and what we see is that the luminal subtype is very well separated from three other subtypes again PC plot but these are the subtypes are relatively close to each other in the first plane principal component one versus two we set number of principal components to be drawn to three that is why we also have planes associated with principal components one and three two and three and here in the next plane you see that cloud and low subtype is well also separated
from basal and normal like but basil and normal like subtypes look similar anyway it please pay your attention to the fact that initially we knew nothing about the subtypes but this analysis provided us an unsupervised but very clear very definitely raishin enter at least two groups but this is a visual inspection and visual inspection can hardly tell us what is a normal well an acceptable a good number of clusters of groups for a given set of samples so here we see that at least two but if we knew nothing about true subclasses we could not
set the number of subclasses 2 3 4 5 so on based just on our visual inspection so let us create one more pipeline for the unsupervised analysis of the data it's just in the same section for performing formal clustering in our case hierarchical clustering of the samples we have so again we go to the unsupervised analysis section again we upload the file expression data this time we do not need marks we don't need information about subtypes about classes that's why we again upload the file cell lines expression data so again it may take some time
we start creating pipeline and this time we want to perform clustering for example using each class using hiragana clustering model so each cost here it is here it is here let me know if everybody's here as here we'll set a bit different options for example we'll set the expected number of clusters to 4 if you like 5 more please keep 5 absolutely not a problem let us change linkage type for example towards d2 so it's more important because linkage type is an essential parameter of hierarchical clustering so linkage type is changed to for example words
d2 definitely it's not the only possible option and that's all that's all again do transpose simply tells which information goes by cones and by rows that's it that's it so we give any name for the pipeline and start it great so while it's running let's briefly discuss what is performed by these pipeline so initially we know nothing about the number of groups so initially we look at our samples which are here illustrated by dots in a plane and associate one group with each individual sample so initially in the bottom-up approach of hierarchical clustering we have
the number of clusters of potential clusters of potential groups equal to the number of samples then we simply unite two samples which are the closest into one group and treat these samples as an inseparable well met a sample acquires a sample so for us currently it's one meta sample but not two individual samples so it's one dot instead of two dots and then we continue the same process we look at two samples of meta samples which are most close to each other and unite them into one group so at each step the number of groups
is reduced by one we continue the process and we unite the closest pair of samples into one groups but here well initially we need to set distance between individual samples and here we need to define what is the distance between meta samples between what is the distance for the situation when at least one argument is not a standard sample but a group of samples well quite a sample so this distance can be defined in two very different ways minimal distance between samples from the first group and from the second group maximum distance mean distance or
more complex definition so it is the question so it's the fact determined by the linkage type how to define the distance between these quasi groups what formula is used so this process goes on and on and on at each step we add we reduce the number of groups by one and finish this process by one group that unites all the samples at this visualization it's hard to understand the logic it's hard to understand how the process goes on but there's once more standard visualization for the out of the hierarchical clustering so-called dendrogram so let us
have a look at the progress of our pipeline well let's inspect what's going on there so the mine is done so please let me know if yours is done as well so if it is again if we want to inspect our results visually we have a PDF file cell lines expression data series each class dot PDF and here you see the results samples in our case breast cancer cell lines go into the line below and step-by-step these cell lines are united in two groups the hate at which two sub groups are united shares the measure
of dissimilarity between these two groups for example if the if grouping is performed at a low height then the united groups are similar if we unite two groups at high height it means that groups are not similar at all so these figure clearly states that we have two well-defined clusters and potentially both clusters can be further divided into two subgroups but if you have a look a close look at this figure you'll see that these groups would just coincide with the subtypes we have for example one of our subtypes normal-like subtype yeah normal like subtype
is located in the rectangle you see marked as green so if we cut this tree into two groups we need to select the cut height at the level of 300 400 we have two groups if we cut the tree at a low height we get four groups if we continue reducing the cutoff height we get more groups but please take a look what will be the next separation if we reduce the height to the level of approximately 200 here you'll see that HCC 202 cell lion will be identified as a separate cluster as a separate
outlier so just the same cell line that was identified by the analysis explained by Julia anyway four clusters I seen really clearly in this figure and there's no wonder that luminal subtype here is subdivided into two classes as indeed it includes two subtypes of the second-level luminal a subtype and women will be subtype we obtain this division into clusters in an unsupervised manner looking at all genes looking at all genes after very single tree filtration exclusion of jeans with concentrate low expression level the question is can we get more looking at lower number of genes
so we can we get more information looking at much lower number of genes it seems that we lose many information limiting our focus on just a few genes but at the same time if we analyze large of genes the majority of genes the majority of transcripts simply introduce the noise so there are only some genes related to the question of interest and reducing the number of genes were looking at can potentially improve signal-to-noise ratio indeed we'll lose some information but still we can keep much information and greatly reduce the level of noise for example let
us look what can be done just in a similar analysis type if we limit our attention on 15 genes out of our initial set so let us create two pipelines similar to the ones we just created just the same types of the analysis but limited to 15 genes again we're in the unsupervised analysis section so first for the PCA analysis we upload the file cell Lyons 15 genes marked in order to see the complete figure cell lines 15 genes marked here it is it's small there are only 15 genes not 7,000 so PC draw analysis
you see a draw analysis is the the first pipeline we're going to create half the second pipeline we're going to create in a very similar way is pipeline for each cluster in this case we upload non marked file cell lines 15 genes cell and 15 genes and again perform job the same steps we upload this small file we upload this small file and create a pipeline each class for example with 4 clusters again you can keep any other number of clusters as it does not affect it does not affect the dendrogram we get and again
linkage type the algorithm for getting distance between coitus samples set to worth d2 we save it and go on and complete this pipeline each cluster and run it okay we'll soon have the results for these pipelines but in any case if we look at these tables will again see that for these tables just a moment let me show you for these tables the selected genes are identified by their in symbol IDs so information that's potentially hardly interpretable as only few of us remember the association of specific genes with their own sample IDs so if we
wants to get more meaningful information about genes gene names let us run the third pipeline and then just check generate the results just gather the results the pipeline we're going to run the next pipeline is associated with the utility section we simply want to know what are the genes were working with so we upload our file cell lions 15 genes the Lions 15 genes and use annotation model that will look at the database and add standard gene symbol and categories to each gene we have in our table so that's it annotation for example with the
default categories okay okay so let me just repeat all the steps the question is how to restore how to restore gene symbols and go-go categories heaven for example in sample gene IDs well technically it can be done simply by using the platform by creating a pipeline at the utility section so we upload a file with genes for which we want to know these gene symbols these annotations just the file were working with and create a pipeline with the annotation model it says that for example gene on transcript IDs located at column one in a table
that has a header we want to add information including GU categories so that is very simple pipeline that's it okay and let us look at the results let us look at the results what we get which pipelines are completed for you which are not in any case what we have for these 15 genes for these 15 genes we have a much clearer separation of groups on the PCA plane so is the figure you get out of the PCA analysis and indeed we see that going into lower number of genes well into the subset of genes
we can call a gene signature we can have clear a figure of what's going on and just a moment and if we see if we look at the results of irrigator clustering you obtained okay perfect so if you download the results of the hiragana clustering you'll see the dendrogram I am having here with additional marks so I just point your attention to the fact that these 15 genes indeed give us clustering into four clusters that perfectly coincides with the division into four subtypes number of genes with an increase of signal-to-noise ratio or an analog of
this signal-to-noise ratio gives us the result perfectly concordant with what we're dealing with so that you record your clustering and finally we're waiting for the results of the annotation which will also give us gene symbols and all the other data type but it is the result of the unsupervised analysis it is the result of the unsupervised analysis definitely there are multiple other methods for unsupervised analysis for example for clustering well known method is k-means there are other methods for clustering that may produce results different from the hierarchical clustering and that may require different input for
example 40 means we do not have a clear visualization with a dendrogram and have two predefined K number of clusters number of groups [Music] in advance but this initial analysis linked with clinical data linked with Vernon typic data gives us separation of our samples into groups and we have another task having the separation of samples into groups how to identify the group for new samples for example having our data set as a learning set we identified that breast cancer patients can be divided divided into subgroups and how to define how to understand which what is
the subgroup for a new patient it's the question for the supervised analysis so probably at this point we may have a short break well if you are a bit tired and finally in our final part before a short hand hands-on practice will briefly describe discuss the analysis in a supervised problem statement when we initially have this division in the subgroups that we could identify using unsupervised analysis okay so as I understand in about 15 minutes okay so in this case we can simply try to have a simple example which will show you that indeed these
methods of the analysis can simply be used to analyze a data set you know nothing about initially so what I'm talking about is the following in the set of files do you have there's a subfolder hands-on what is this set of data we have in this file we took two additional projects both dealing with breast cancer but in one project the samples were human samples just biopsy samples their biopsy samples or tissues taken in frames of surgeries and the other data set is Centegra is data originated from a patient arrives and graft project so human
breast cancer was implanted into mice and then taken again out of the non-human mice micro environment and it's clear that micro environment in human and in mice should be different and disabled samples should also be different so we took just the same 15 genes to make the analysis faster but the samples here are intermixed so some samples are associated with humans Sam associated with mice so could you divide this set into a human set and my set probably not named in each one but telling which samples belong to the first and which samples belong to
the second using the automatic modules we've discussed so in order to understand that this analysis is really simple in spite of in spite of the fact that the number of the amount of information is relatively large while dozens of samples and several genes so the task is to separate these datasets into two different really different subsets mice and human derived samples this the task clear so as the heel is walking there behind said he'll just let us know if everybody is following if everybody has seen the file it's in the original zipped folder that you
download it there is a file fill hands on and the only file in the folder hands on file with intermixed file with intermixed datasets human derived and mice do ok and people that are just joining online you can also let us know on chat if you are following along or if you are having any issues in the meantime I just want to mention also Vladimir there is somebody requesting clarification on the steps for the annotation pipeline so if at some point we can review that again that would help sure sure so probably a good option
is to give several minutes to implement these hands on task and then return to the annotation pipeline okay if you've started your pipelines and just waiting for the results well please let me know and we can briefly discuss these annotation and then to go through some additional topics very briefly [Music] [Music] okay so again regarding annotation here currently we're talking about very simple annotation about taking an assembled gene or transcript IDs and add an information which is simpler for manual interpretation i mean gene symbols so just standard gene names and gene ontology terms cell localization
gene ontology functions and so on and in order to perform this pipeline definitely all we need is the set of example gene or transcript identifiers in our case these identifiers allocated into in the first column of the table so we simply upload this file we simply upload this file but this one for example and tell that we need to add information for identifiers stored in the first column of the table that the table has a header so the first row is not it is simply a header and we want to include gene ontology data into
the output that is all that is all and the output for this pipeline I hope you also have an output for this pipeline includes a table marked with out suffix so it is just an initial table in the week in the right gene symbols are added so here for these assembled genes gene symbols are added and here you see that in our subset of 15 genes there is for example a very well known epithelium cell adhesion molecule he become is present and then goo terms are associated with each gene for example for the case of
this well-known hidden molecule Rahzel domes include plasma membrane as its adhesion lateral plasma membrane integral component of membrane and others so for for example DC and prodding these Gautam's include wound healing as a biological process along with gene symbols here we see food well a bit fuller description full gene name for example collagen type 6 primer etc so it's a simple very simple annotation limited to add in gene symbols gene ontology annotation so if you are ready with the hands-on test we can discuss the results and then briefly discuss just a couple of more topics
whatever there is also a question here if you will after this hands-on work go over a standard differential expression pipeline with four annotation exactly exactly so again and asking Sahil is the voice as the voice from there what are the approaches you test it in order to separate the data set into two subsets and what results did you get any ideas you're also welcome to write your ideas on chat and we'll announce them here okay so both options we discuss to the principal component analysis and hierarchical clustering analysis both of them can solve the problem
we have here for example for example if we perform here our kacal clustering for this file well for example I'll show you the result with two factors in an output well we'll see what's happening and principal component analysis potentially can also call this task but the output will be a bit longer to interpret so initially we have groups completely intermix okay the answer here from the audience somebody said PCA with two groups exactly so PC give two groups let me just take a look okay for example as we are waiting for the result I'll run
this pipeline and it's really fast [Music] so what we have for hierarchical clustering as an output here we see two well-defined groups and the answer for this shot for this simple task is the following all samples marked with odd numbers belong to one group all the samples marked with even numbers 256 60 etcetera belong to two groups so this separation is just because indeed one subset is human derived and the other subset is mize derived and in human derived samples all the samples belong to the same subtype so that's why the unification height is relatively
low and in the my stride subset in PDX subset there were two types of breast cancer and these two types are defined by these clusters that can be obtained by cutting the tree at the height of for example 30 so indeed again here occur clustering gives us a clear separation and if we look at the results of the principal component analysis after we get it it's it will take just or seconds we'll also see these clear separation for the hierarchical clustering definitely we have not only the plots but we have additional information for example we
have a file with the suffix clusters only dot txt and just a moment what is the content of this file I'll open it in Excel to a simpler view so for each sample this file gives a cluster identifier we set the number of clusters equal to two so the cut off height was selected in such a way that the result a number of clusters is two if we've said the cut off level two three four etc we'll have the same number of clusters here and looking at samples united in one cluster is very simple we
just look at samples marked with the same cluster ID and in the pc plane we indeed also have a similar separation into two groups one group the left and bottom and the other group top right so it is the separation so having groups heaven the groups we can think about the next problems and one of the problems is to find genes that have different expression in these subgroups we're dealing with so first we need to we need to define we need to analyze to understand what does it mean a gene has different expressions in the
in to compared classes in to compared subgroups first we may look at mean expression levels the different expression means that average expression levels are also different but look at the example features to the right of this slide you definitely see no differential expression in the upper case and evident differential expression in the lower case in that blue group of samples expression level is evidently much higher than in the red group of patients at the same time the difference between means in the upper and antilochus is just the same one in log to scale so two-fold
difference in the initial scale that is why fourth change the difference between means can be used as the only parameter as the only characteristic of differential expression we also need to consider statistical significance of this differential expression obtained using one of the standard or non-standard statistical tests in the simplest case it is a student test or so called T tests that compares the difference between average levels of expression levels compared to deviations compared to variations inside expression levels in each group more complex cases which are better for small sets of samples use more complex statistical
schemes for example statistical schemes based on negative binomial distributions are popular technically in order to get the set express genes if we want to use a simple test for example t-test we can do it even taking expression levels provided by our previous pipeline one of our previous pipelines as an input but more complete as I told more complex statistical approaches are preferable so in order to use more complex statistical approaches let me just show you the pipeline design we can create this pipeline together all we need is to upload samples where it wants to compare
my case I'll use another set of links to files that are already present in the platform links cloud in low versus normal like samples the above six samples in this set are close in low breast cancer cell line samples and lower 5 samples are normal like samples but in these case so in this case we want to compare expression levels in true route to want to find genes that have higher expression in one group and lower expression in the other group so we need to specify which samples belong to which group that potentially can be
more than two groups but let us limit our consideration to two groups so for example as I told in these case in these case lure fire samples belong to one group and above six samples belong to the other group and if we initially know the difference between which sample belong to which group here we press Continue as we have groups well submit changes in groups and create a pipeline similar to the ones we initially had as here we again start with raw sequencing data with first you data so I just recreate the pipeline we had
initially but here at the end we wants to find the set of differentially expressed genes of differentially expressed genes it can be performed if we want to use more complex statistical models based on negative binomial distribution for example by well known H R or D SEC two modules so technically technically that is all so we give a name pipeline and run it regardless of the module will we're using regardless of the module we're using what we get as an output what we get as an output annotated output for this pipelines is provided in the sub
folder output files then we can go further into the differential expression subfolder so the outputs have similar meanings and I'll pay here attention to the main information just a moment sorry just a moment so let us take any what in what information can be used here so let me sorry just a moment just a moment okay okay so what information we have after the differential expression analysis for each gene or for each transcript we have full change potential mainly log for change the science shows in which group the expression level is higher then we have
statistical significance of the differential expression the probability that we'll have the same or even higher for change in the case when in reality there's no difference in mean expression levels adjusted p-value which is the main parameter estimate that is examined if we want to look at the set of really differentially expressed genes and the base level so the mean expression level in both groups or the main expression level in just one of the groups it depends on the instrument by using so if the standard output of n a 2 for the analysis of differential expression
so we'll get just this similar we'll get just a similar output for dc-to-dc HR etc and what our first do you know what is the reason for adjusting statistical significance for adjusting p-value in this case so why do we need to additionally add some adjusted p-value which is also denoted as a family false discovery rate if they are proven to value etc so the reason is the following it be explained by a very simple example imagine that you have a coin you drop this coin ten times and ten times it fells with the same side
up so it is suspicious the probability that it's a standard coin and it dropped ten times all ten times the same side up the probability is low but if we repeat this experiment for several thousand times we'll definitely have several coins with this behavior it does not mean that these coins are not normal it potentially simply means that the number of tests we were simultaneously writing the number of points was too high and what we see is associated not with individual coins but with the number of tests so this adjustment takes into account the number
of tests the number of genes we're dealing with and in order to select differentially expressed genes in a standard sense so what are the standard thresholds standardly a gene is called differentially expressed if adjusted p value to value FDR does not exceed 0.05 that is 5% and standardly has full change of at least two times so log full change of at least one so higher than 100 with n minus 1 these are the thresholds that are not universal in different types of the analysis must treat thresholds can be used for example 0.01 one to stand
for adjusted p-value and more severe threshold for the full change additionally in many cases lower express genes are simply eliminated from the analysis as we initially discussed that the error in quantification of expression levels for these genes is much higher but anyway the result of this analysis is the table which further allows you to select main players in the field of differentially expressed genes so the main parameters are full change statistical significance mainly measured in adjusted prevail your false discovery rate as another name for the same and additionally it's a good idea to exclude genes
with low expression after having genes after having this analysis we may think about further biological interpretation we may try to think about the following question is there any pathway is there any pathway is there any well experimental group of genes are expressed by differentially expressed genes can we say that in our experiment genes associated with a particular biological sense increase or decrease their expression and there are multiple approaches to try to give an answer to this question of interest so the first idea is very simple and it is utilized for example by the well known
delete online service david service for gene enrichment analysis let us simply take a set of genes with the highest level of differential expression with the lowest adjusted p-values and highest for changes and simply forget about these additional parameters well let us simply take these genes and let us try to lies whether these top jeans well say 100 or 1000 genes are enriched by genes belonging to a particular pathway what does it mean gene this gene set is enriched by genes belonging to a particular pathway it means that if we take random genes with a number
equal to the number of genes in our pathway the probability of getting the same number of genes belonging buthe to the pathway and to power a set of differential expressions is low so for example in our set we have 50 genes belonging to this pathway and in reality if we take the same number of genes the probability of having 50 or more genes is low so that is the approach used for example by diet as the first generation gene enrichment tools the problem here is the following we ignore too much information with not too much
information associated with genes in the results of differential expression analysis for example we completely forget we completely forget about the level of differential expression for example about full changes here losing such a great amount of information leads to poor example at the poor results of the analysis the second level of these instruments takes into account these additional information about differentially expressed genes for example log fold changes and we have a platform for example there's a module based on our our script gauge adopted for a simple use with human experiments and the gives us an opportunity
to give a national interpretation to give a natural interpretation of the differential expression analysis results so in order to save time I'll just show you what we get in the enrichment analysis for this experiment first just a moment first for each analyzed pathway we get a statistical significance of enrichment so we see which pathways are really enriched by genes in our set of differentially expressed genes sandal parameter used is adjusted devalue that in many cases denoted as Q value so here we see that for example focal adhesion get some signal pathway our Y differentially expressed
genes and if we're looking at standards pathways for example KEGG pathways we also have a national visualization of differential expression we have pathways we have pathway in which players are colored and the color code is based on the full change level genes differentially expressed in one direction for example downregulated are marked in one color and genes with the opposite regulation are marked with another color so this analysis is non-trivial from the statistical point of view but it is trivial from the technical point of view using the platform so all we need to do is to
upload the file but here please take into mind that for these type of the analysis if we want to use the complete information about full changes we need to upload the hell table not only differentially expressed genes but the whole totality of genes in the experiment as the method requires all information as the background for its statistical model and the output is just what I'm showing you so technically this unfree Ville analysis can be performed simply in the case of Talbert by and for platform in the utility section by using the human gauged model unfortunately
it seems that the time is coming to its end but you've got all the files for testing it and I definitely will be happy to answer your questions if you'll try to test it and if you see that certain parameters seem to be unclear so you have a form here to present your questions and we'll definitely try to answer them so yeah as far as I see problem where I'm out of time and we will be happy to try to answer your questions but also I you can let us know I know that some people
had to leave if you don't have to leave we can just finalize this one last pipeline and look at the outputs if you would like to there's a few more things to that pipeline that could be shown but you you can let us know I don't know so he'll if you can okay so that somebody is asking is it possible to save the file after adapter cleaning RNA editing run okay it's it's simple question and I'll give a fast answer to this question if we look at the pipeline any pipe landing with these modules we
have final results in the main download section and the main download section but all intermediate results are also well I am just looking at the complete pipeline in my stack just a moment example hopefully here it is yeah so all intermediate results are also accessible in the folder job stem profiles so for each job for example tremor Matic BCR cleaning there are all intermediate file saved and they're also downloadable so all statistics all intermediate files are accessible ok so that answers that question so the question about what the pipeline looks like with gauge with it
with the enrichment analysis we we can finish that pipeline and just look a little bit more in detail into the outputs of the pathways if everybody has time so if you have time let's just continue we'll take just a few more minutes Vladimir do you want to just demonstrate the pipeline with gauge and okay just try to find files we have okay it's absolute not a problem so again let us briefly take first a look at the input for this pipeline the gauge may deals only with two types of gene identifiers gene symbols and and
tres IDs so insane bill identifies should be transferred to something like that it can be done simply with the annotation section probably it will require some time to annotate thousands of genes but in any case it's not a problem so what is the mood for the gauge pipeline it is just the output of the differential expression analysis pipeline so we have IDs we have the parameters that characterize the rate of differential expression the gauge takes one parameter as an input standardly it's log to full change and additionally as I said gauge deals with gene symbols
or interest IDs so we added gene symbols using sample IDs that were initially present in the output so is the input the minimal input and in fact the complete input that is used contains two columns gene identifiers given by gene symbols or interest IDs and rates of differential expression given for example with log two for changes that is the input and in order to generate this pipeline we have to go to the utilities section upload this file for the deseg two out this is a file district full output annotated annotated here means that we just
edit gene symbols from the HR a simple file is one below there we have just a slightly different of columns so I download I upload this file to the system and let us take a look at options we need to specify for this model the options are quite simple and comments are accessible if we we point at the I section we have explanations in our table let me show it once again we have the header so corn headers are present corn with gene IDs where gene IDs are gene symbol or entrez ID in our case
is the second one and let us remember that full change is allocated in the fourth column so gene identifier is allocated in the second column second column sorry bring characteristics of the french expression for example full changes are located in to the fourth column there are two types of the analysis parametric or nonparametric both are applicable to fall changes if we use statistical significance cout value for example as a matter of differential expression nonparametric statistical approach is preferable then we select the set of genes the set of pathways we are going to look at for
example standards get met sig is the set of KEGG pathways related to metastatic cakes metastatic and signaling pathways finally we set a direction of differential expression that is taken into account combined means that we combine up and down regulated genes separate means that we separately annal analyze downregulated genes and upregulated genes and down and up means that we limit our analysis only to down or up regulated genes now the final threshold is simply for drawing will draw the best ways with enrichment significance that does not exceed threshold of 10% so false discovery rate below 10%
and that is all so that is all we need for this pipeline that is all we need for this pipeline it will take some time to be completed as the input here as I said completes the complete set of genes here we left even unexpressed in so 60,000 but it won't take too long so probably if you have some additional questions we'll see the output just today ok so additional questions one is can you draw a curve of the gene set enrichment analysis as an output from this gauge but so here I need to understand
what the meaning by the curve associated with gene and Reena I can I have this question from Andy B I don't know if you're in the audience maybe you can ask directly yes so if you just provide a comment what curve do you mean I'll give you an answer so currently we have all these pathways cake pathways Stanley their allies get pathways with color codes of differential expression and the text tables so currently additional curves are included but in the majority of cases it's not a problem in any way for multiple modules including this one
we output not only the results of the processing but we also output our scripts that are used in our inside the platform to provide the analysis results so what we also have is an R script that performs everything from taking initial data to producing the plots so if any additional information is required this R scripts can be used as a base and it can be slightly edited for example by user if a user knows bases of coding to get any additional information available inside and inside this type of the analysis another in what situations do
you supervise and in what situation to use unsupervised okay thank you very much for this question the difference is in problem statements so these methods provide answers to completely different questions in the first case we know nothing about separation of our samples if we analyze samples in the subgroups so we just have a set of samples and the task is to understand whether this group of samples can be subdivided into any well more or less natural subgroups we do not know this division but in this type of problem statement we try to find this division
so in any case unsupervised analysis means that we know that we have no additional information about these samples in the supervised analysis we initially have information about samples for example about classes of samples about subgroups of sample and the question is not to divide the group of samples into new subgroups we already know the subgroups but the question is the following how can we construct 2yoon a classifier function that will take new samples as an input and attribute these new samples to one of the groups we initially had so we had patients we already divided
them somehow into subgroups and then for new patients we wants to attribute these new patients to one of the groups we initially had this is the problem for the supervised analysis type okay with order could you please repeat your question okay sorry so what about replicates okay near analyze biological areas well it depends again it depends on the problem we're dealing with in our case it depends on the experiment you're working with I won't give you the universal answer here but in a way in many analysis biological replicates can be just be grouped into one
subgroup a group of samples in the analysis so we can account this information that a set of what a number of samples are simply biological replicates by United these samples enter subgroup but again a lot depends on the design of the experiment of the number of major groups we have number of different conditions we have we definitely should understand that there are independent samples there are biological replicates for some well conditions for some effects and moreover in multiple experiment designs there are technical experiment technical replicates we definitely should take this into account and do not
treat technical replicates similarly to treating independent samples but unfortunately I don't know the universal answer here it depends again on the macro design of the experiment okay we have enough yeah can the platform perform whole exome analysis just a moment try to give an answer usually yeah it depends on the goal of the analysis the answer is yes for the many types of the health exam analysis for example for the identification of snps of copy number variation there's a section that performs this job currently we're trying to update this section to add some modern approaches
but the formal answer to this question is yes there's such a section what was that I did not hear that are their genomes non-human genomes so basically we have several model organisms here so you see the list starting from Earth adoptions finishing with well relatively rare types of genomes it's not complete these are just the genomes we had to work with but adding a new genome is a simple relatively simple task so if there's a project or there's a user who needs to deal with another standard genome is genome can be added to the system
and edits to all these forms currently I see that the guiche pipeline is completed so as we discussed the output here is gathered in one zip archive just in order to avoid that multiple files and the type of the market the type of files here is the one we discussed we have a simple set of pathways with enrichment statistical significance associated with each pathway and visualization for enriched pathways okay so just to summarize I know that people have to leave but just to summarize we were able to go through several sections next-generation sequencing data pre-processing
including trimming technical sequences and removing PCR duplicates RNA seek based quantification of expression levels so we looked at standard mapping pipelines we did touch on novel isoforms not in detail as we hope to but we got through a lot of it we also looked at gene set enrichment analysis and this is just the output here you can see that genes are annotated with gene ontology and gene descriptions as well as you can get all the way to pathway and wait and see which genes within the pathways are under its plaster over expressed as well as
we looked at some of the machine learning methods analyzing expression data we looked at unsupervised methods like PCA and clustering techniques and then we looked at how to build a supervised analysis using such methods as SVM and so I get to mention the data that we used we use data from two publications this whole line based modeling of precision technology answer a project that is available on our educational site as well as whole transcriptome profiling of pdx models as a tool to identify both tumor and stromal specific biomarkers and you can find the link to
this publication as well and it's also a project that is available on our educational site that you can play around with and finally thank you for dr. glen america ladunka who is the chief scientist at the tower by informatics Research Center and Julia fell off who is the PhD student that uses the platform in some of our research projects and again special thank you to dr. Judy Iqbal from UNM see I have to organize the room and brought a lot of the people to the workshop and we will be staying in touch by email if
you want to email us to heal myself or schedule a time we plan to be there again and organizing another workshop if you are not able to attend or if somebody that you think should attend a workshop like this please let them know on the 20th we will have another workshop at UNL and in Lincoln asks a heal about that workshop it will cover most of the same steps that we did today but we will also touch on some new things coming Thank You Leah
Related Videos

59:35
Machine Learning for Biomedical Data: Free...
OmicsLogic
1,433 views

14:22
Transcriptomics 1: analyzing RNA-seq data ...
OmicsLogic
39,931 views

50:46
RNA-seq Analysis 2023 | 01: Introduction t...
Bioinformatics DotCa
1,931 views

51:48
Illumina Sequencing Overview: Library Prep...
Ambry Genetics
176,861 views

11:21
Introduction to Weighted Gene Co-expressio...
Bioinformagician
24,089 views
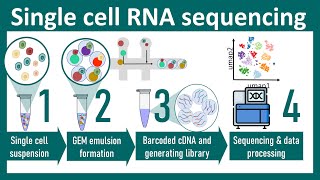
17:41
Single cell RNA sequencing overview | ScRN...
Animated biology With arpan
14,840 views

57:35
How to analyze RNA-Seq data? Find differen...
Candice Chu, DVM, PhD, DACVP
216,847 views

36:09
The Beginner's Guide to RNA-Seq - #Researc...
Applied Biological Materials - abm
141,372 views

1:18:40
Complete single-cell RNAseq analysis walkt...
Sanbomics
81,351 views
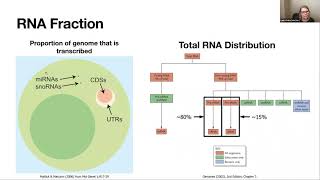
58:58
Webinar #11 - Beginner's guide to bulk RNA...
opar
15,253 views

53:55
Kipoi Seminar - David Kelley (Calico)
Kipoi Seminar
1,140 views

31:11
Introduction to RNA-Seq for Researchers
Iowa Institute of Human Genetics
96,023 views

1:15:44
Bioinformatics for RNAseq
Tufts Technology Services
26,406 views

1:00:23
A Beginner's Guide to RNA-Seq
Novogene
8,771 views

28:02
edgeR: differential analysis of sequence r...
Bioconductor
8,107 views

1:02:18
How to Read a Cancer Genome | Part 1: The ...
Genomics Education Programme
9,576 views

31:50
Setup RNA-Seq Pipeline from scratch: fastq...
Bioinformagician
95,864 views

18:26
StatQuest: A gentle introduction to RNA-seq
StatQuest with Josh Starmer
494,588 views

7:40
Microarrays vs RNA Sequencing
LC Sciences
115,194 views